 网站地图
网站地图

 下载:
下载:




-
从分子或原子的水平上,实时观测、跟踪化学反应过程,最终实现对化学反应的控制和操控,是化学家们一直以来追求的目标。人们对化学反应机理与历程的研究不断地取得了新的突破。1935年,EYRING等人提出了过渡态理论[1],认为反应物变成产物之前,要经过一个过渡态,即:反应物→过渡态→产物。由于过渡态是一个寿命极短的态,寿命一般大约为10-15s的数量级,常规的实验方法对寿命极短的过渡态实时观测与分析几乎不可能。因此,长时间以来,在化学反应的途径上所经历的这个过渡态只是一个理论的假设,成了一时间打不开的“黑匣子”。
为了能够实时跟踪原子与分子内部动力学信息,了解反应物转变为产物过程中存在时间极短的过渡态,美国物理学家ZEWAIL教授于20世纪70年代起开始尝试把短脉冲激光应用在分子动力学过程的研究中。到了20世纪80年代,随着飞秒激光器的成功研制,使得实验上观察过渡态成为可能。ZEWAIL教授把飞秒激光技术应用于分子反应动力学过程的研究中[2-4],实验上成功探测到了化学反应中存在的寿命极短的过渡态。ZEWAIL教授因此获得1999年诺贝尔化学奖。
在超短脉冲激光出现以前[5],人们常通过一些间接手段推断激发态的寿命。如由于影响光谱线宽的很多因素中,仪器加宽、分子碰撞加宽等的影响相对比较小,可以被忽略,而寿命引起的均匀谱线加宽的贡献最大。如这种假成立,通过测量吸收光谱的线宽,理论上可以得到激发态寿命。但为了避免自然寿命以外的其它因素对光谱加宽的影响,从激发光谱中得到自然线宽,必须满足以下几个条件:(1)自然线宽必须大于激发光束的光谱宽度;(2)必须为谱线设定一个特定的线型,这样才能测定确切的线宽;(3)多普勒加宽、压力加宽等的影响必须预先被修正或被排除;这种理想的情况即使利用特殊的技术,由于考虑到以上众多的因素,在实际的实验测量中也很难实现。由此可见,在实际实验测量中通过自然线宽来准确测量激发态的寿命很有难度。但这种方法的优点是在大多数情况下可以很容易获得激发态寿命的下限。除了自然线宽中得到激发态寿命的下线以外,还有一些其它的间接方法也能够估算或给出激发态寿命的下限,如产物角分布、吸收线宽、喇曼极化等,但时间分辨测量更直接和明确。
飞秒激光技术的出现[6-7]实现了对激发态寿命的直接测量、激发态动力学的实时探测、过渡态的实时跟踪,并能够给出相关动力学过程的清晰的物理图像。飞秒时间分辨质谱技术是飞秒抽运-探测技术与飞行时间质谱技术的结合,利用一束飞秒激光作为抽运光,使分子激发到感兴趣的态上,制备随时间演化的量子波包。另外一束飞秒激光在不同时间延迟下探测量子波包的演化,这样就可以得到时间分辨的飞行时间质谱。由时间分辨质谱可以准确测量分子激发态和里德堡态寿命,根据飞秒时间分辨质谱得到的数据可以推断分子激发态势能面的非绝热耦合信息,以及分子过渡态的信息,因此,在分子激发态和里德堡态的研究中, 飞秒时间分辨质谱技术已成为非常有用的手段。
-
飞秒时间分辨质谱技术的原理如图 1所示。首先运用两束超短飞秒激光脉冲:抽运光将分子从电子基态激发到电子激发态或里德堡态上,制备随时间演化量子波包; 探测光用来电离所制备的激发态或里德堡态上的分子。抽运光和探测光与样品发生作用产生的离子在电场中进行加速,再通过一段自由飞行区最终达到探测器,被光电倍增管(photomultiplier tube,PMT)和示波器所组成的探测器探测。图中S1和S2分别表示分子第一、第二单重激发态,Δt为抽运光和探测光之间的延迟时间。

图 1 飞秒时间分辨质谱技术原理示意图
抽运激光选择不同的激发波长来选择性地激发感兴趣的电子激发态/里德堡态,制备随时间演化量子波包,当分子被抽运激光激发到特定的电子激发态/里德堡态后,会通过不同的通道发生衰减,探测激光在不同的抽运探测延迟时间内探测分子的衰减过程。这些衰减过程一般都发生在皮秒甚至在飞秒的时间尺度内,为了能够实时跟踪激发态超快衰减过程,并观测激发态在极短的时间内发生的动力学过程,抽运激光脉冲和探测激光脉冲之间要有能够控制在皮秒甚至飞秒时间范围内的一定的延迟时间Δt。随着精密位移平台技术的发展,时间延迟控制在几个飞秒的时间精度成为可能。目前通过精密位移平台可以很容易达到很高精度的位移扫描。因此,飞秒时间分辨的实验中通过精密位移平台技术精密控制抽运光和探测光之间的光程差来实现飞秒或皮秒量级的时间延迟。
抽运光、探测光和样品分子发生作用产生离子以后,产生的离子或电子通过精细设计的极板产生的电场进行加速。加速离子的电场一般由几块极板所组成,在极板上加电压后,极板有一定的电场强度。第1块极板加较高的电压,接下来的极板加比前面一块极板较小的电压,产生拉出场和加速场,将离子从反应区域中拉出并对离子进行加速。在外电场中加速后,各种离子都获得一定的动能E:
$ E = m{v^2}/2 $
(1) 由此可见,在外电场的作用下, 加速后的质量m不同的离子会获得不同的速率v。因此,质量不同的离子以不同初速度进入无场区进行自由飞行:
$ v = \sqrt {2E/m} $
(2) 质量不同的离子从进入长度为L的无场自由飞行区域, 到达探测器的自由飞行时间为:
$ T = L/v = L\sqrt {m/\left( {2E} \right)} $
(3) 由此可见,离子达到探测器的时间(离子在探测器上出现的时间)T和m1/2成正比。具有特定能量E,而荷质比m/z(m表示带电体的质量,z表示带电体的电荷量)不同的离子在外电场作用下被加速后,会获得不同的速度。由于质量不同的离子以不同的初速度进入无场自由飞行区域,经过一定长度的无场自由飞行区飞行之后,前后达到离子接收器,在不同时间被探测器探测。因此,时间分辨质谱测量不同离子的飞行时间,可以分辨不同的离子。而通过改变抽运激光脉冲和探测激光脉冲之间的延迟,记录同一个离子不同抽运探测时间延迟下的质谱,离子在不同抽运探测时间延迟下的质谱反映激发态布居数随时间演化。
在飞秒时间分辨质谱实验中,由于激光脉冲的零点和飞行时间零点之间可能存在差异、激光脉冲具有一定的宽度,这些因素都会影响分子飞行时间的测量。考虑到这些因素对实验结果的影响,对未知分子进行飞行时间测量之前,必须对飞行时间进行校准。实验中,通常首先对已知分子的飞行时间进行测量,参考已知离子的飞行时间对飞行时间进行校准,再对未知分子进行测量,并对未知分子进行准确标定。校准公式如下:
$ T = a{m^{1/2}} + b $
(4) 式中,a和b是未知参量,需要先求出未知参量的值来得到校准公式。因此,在实验中,通常把两个或两个以上已知离子飞行时间T和m1/2的数据代入(4)式得到未知参量a和b,然后根据实验中测得的未知离子的飞行时间求出未知离子质量。
飞秒时间分辨质谱实验中的质谱分辨率是一个非常重要的参量,飞秒时间分辨质谱的分辨率M可以用以下公式表示:
$ m = \left( {2E} \right){\left( {T/L} \right)^2} $
(5) $ \Delta m = \left( {2E} \right)2T\Delta T/{L^2} $
(6) $ M = m/\Delta m = T/\left( {2\Delta T} \right) $
(7) 式中,m和Δm分别是质谱上的离子质量及其半峰全宽,T是离子自由飞行时间, ΔT为离子飞行时间谱线的半峰全宽,L为飞行距离。由此可见,飞秒时间分辨质谱的分辨率是M在数值上的体现, M的数值越小,说明质谱分辨率就越低,反之,质谱分辨率越高。很多因素都有可能影响质谱分辨率。设计质谱仪时,为了达到较高的质谱分辨率,可以尝试使用反射式的飞行时间质谱技术。反射式飞行时间质谱仪通过增加自由飞行距离来增加离子的自由飞行时间T,从而有效提高质谱分辨率。当飞行时间质谱仪已设计好、各部分的几何长度已确定时,没办法改变飞行时间,这时可以改善电场的设计、调节拉出场和加速场的强度比来改善空间聚焦条件,有效提高质谱分辨率。飞秒时间分辨质谱跟其它质谱一样具有分析质量范围宽、收集效率高等优点。
在飞秒时间分辨实验中, 光源飞秒抽运光和飞秒探测光脉冲都是高斯脉冲,具有一定的脉冲宽度,除此之外,实验中用到的光电倍增管等光电转换器件都不可能瞬间响应,都有一定的仪器响应时间,因此,在实验中所记录的不同时刻的离子/电子信号强度Isig为激发态布居数随时间变化的真实信号和抽运光与探测光脉冲的相关函数的卷积。
如果假设分子吸收一个或多个光子激发到激发态A,由于激发态不稳定,被光激发的激发态A会衰减,A态衰减到另外一个激发态B,B态也有可能衰减到C态,第1步的速度常数为k1,第2步的速度常数为k2,这样一个(A→B→C)反应过程的速率方程如下:
$ \frac{{{\rm{d}}A\left( t \right)}}{{{\rm{d}}t}} = - {k_1}A\left( t \right) \Rightarrow A\left( t \right) = A\left( {{t_0}} \right){{\rm{e}}^{ - {k_1}t}} $
(8) $ \begin{array}{*{20}{c}} {\frac{{{\rm{d}}B\left( t \right)}}{{{\rm{d}}t}} = {k_1}A\left( t \right) - {k_2}B\left( t \right) \Rightarrow }\\ {B\left( t \right) = \frac{{{k_1}A\left( {{t_0}} \right)}}{{{k_1} - {k_2}}}\left( {{{\rm{e}}^{ - {k_2}t}} - {{\rm{e}}^{ - {k_1}t}}} \right)} \end{array} $
(9) $ {k_2} = 0 \Rightarrow B\left( t \right) = A\left( {{t_0}} \right)\left( {1 - {{\rm{e}}^{ - {k_1}t}}} \right) $
(10) $ \begin{array}{*{20}{c}} {\frac{{{\rm{d}}C\left( t \right)}}{{{\rm{d}}t}} = - {k_2}B\left( t \right) \Rightarrow C\left( t \right) = \frac{{{k_1}{k_2}}}{{{k_1} - {k_2}}}A\left( {{t_0}} \right) \times }\\ {\left[ {\frac{1}{{{k_2}}}\left( {1 - {{\rm{e}}^{ - {k_2}t}}} \right) - \frac{1}{{{k_1}}}\left( {1 - {{\rm{e}}^{ - {k_1}t}}} \right)} \right]} \end{array} $
(11) 由此可见,激发态布居数随时间变化为一个单指数衰减、单指数上升或单指数衰减加单指数上升函数。在时间分辨质谱实验中,飞秒抽运激光脉冲和探测激光脉冲都是高斯脉冲,由于两个高斯脉冲的相关函数仍然是一个高斯函数[8],飞秒抽运光和探测光的相关函数仍然为一个高斯函数。因此,实验中先测得抽运光和探测光的相关函数,再由测得的不同抽运探测时间延迟下的质谱信号通过指数函数和高斯函数的卷积公式进行拟合,就可以获得分子激发态或里德堡态准确的寿命:
$ \begin{array}{*{20}{c}} {{I_{{\rm{sig}}}} = \sum\limits_i {{A_i}\exp \left( { - t/{\tau _i}} \right)} \otimes }\\ {\frac{1}{{\sigma \sqrt {2{\rm{ \mathsf{ π} }}} }}\exp \left[ { - \frac{{{{\left( {t - {t_0}} \right)}^2}}}{{2{\sigma ^2}}}} \right]} \end{array} $
(12) 式中,Ai是振幅,τi是寿命,σ是相关函数的半峰全宽,t0为零点时间,⊗表示卷积运算。
飞秒时间分辨质谱技术可以测得不同抽运探测时间延迟下分子电子激发态电离或解离而来的离子质谱,由不同抽运探测时间延迟下质谱信号强弱的变化反映激发态布居数随时间变化。从分子激发态布居数随时间变化的信号可反演出分子激发态的动力学信息。因此,时间分辨质谱技术在研究分子电子激发态非绝热动力学过程的研究、里德堡态研究、过渡态研究中发挥着很重要的作用。下面以几个方面介绍飞秒时间分辨质谱在超快动力学中的应用进展。
-
飞秒时间分辨质谱技术通过测量母体离子和不同碎片离子信号随抽运探测时间变化,能够归属分子解离通道,能够观测分子解离的过程。LUDOWISE等人用飞秒时间分辨的质谱技术研究了OClO分子X2B1态的解离过程[9],他们利用飞秒时间分辨的质谱技术扫描母体离子和各个碎片离子的时间分辨质谱,并对实验数据进行分析,指出OClO分子X2B1态的解离主要是二体解离。最近几年这方面比较典型的工作是台湾清华大学ZHENG教授团队利用时间分辨的质谱技术对二甲基亚砜(dimethyl sulfoxide,DMSO)[10-11]、丙酮[12-13]等分子电子激发态的超快解离过程进行了详细的研究, 并取得了突破性的进展。
图 2是为ZHENG教授团队用时间分辨质谱和理论模拟相结合,对二甲基亚砜电子激发态超快协同解离动力学过程进行研究的实验结果,以及理论模拟的结果,这个工作也是飞秒时间分辨方法对协同解离动力学过程的最详细研究。他们利用200nm飞秒紫外光将母体分子激发到第二电子激发态,由于二甲基亚砜分子的第二激发态是解离态,被激发后会发生快速解离。二甲基亚砜分子第二电子激发态的解离存在多个解离通道,会产生多个碎片离子,选择合适波长的探测激光分别对母体离子和各个碎片离子进行共振探测,可以得到母体离子和碎片离子的飞秒时间分辨质谱。实验中观察到了解离而来的自由基SO,并记录SO自由基的时间分辨质谱。二氯亚砜母体分子解离时两个甲基都断掉才会出现到自由基SO,SO自由基的出现反映了二氯亚砜的解离通道中存在两个甲基都断掉的解离通道,而SO强度随抽运探测时间变化(飞秒时间分辨质谱)反映了自由基SO的生成和衰减过程,从SO自由基的飞秒时间分辨质谱中,可以推断处于激发态的二甲基亚砜分子如何断掉两个甲基进行解离。他们用实验和理论相结合观察到了产生自由基SO的多个通道。最有意义的是,观察到了二甲基亚砜分子产生自由基SO的快速通道,这个快速通道的时间尺度τ1=50fs,他们解释50fs的快速解离通道为二甲基亚砜分子吸收200nm紫外光子激发到第二电子激发态之后通过快速的协同解离通道进行解离。图中, TMSO指四亚甲基亚砜,R表示键距,R(S—C)表示S—C键距,λp为抽运光的波长,ν″为振动态,τ2为振动态寿命。

图 2 二甲基亚砜电子激发态超快协同解离实验结果和理论模拟
GITZINGER等人利用飞秒时间分辨质谱技术研究了CH3I分子第2个吸收带B带的预解离动力学过程[14],如图 3所示。他们用201.2nm的抽运光把CH3I分子激发到B带,通过扫描母体离子和碘离子时间分辨的质谱信号,观察到了母体离子信号随时间衰减的同时电离子信号随时间增加,因此,得到了CH3I分子B带预解离的有力证据,并给出了预解离时间为1.5ps。
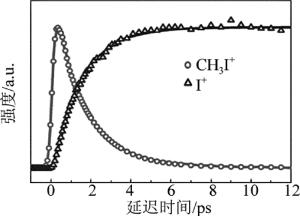
图 3 碘甲烷分子B带超快预解离动力学过程的飞秒时间分辨质谱
中国科学院ZHANG小组利用飞秒时间分辨质谱技术研究了丙烯酸分子的超快预解离动力学[15]; 利用400nm抽运激光、800nm探测激光研究CH3I和C2H5I[16]等分子预解离动力学过程,观察到了这些分子体系超快预解离动力学过程; 利用266nm抽运光、800nm探测光研究了邻碘甲苯分子光解动力学[17]。POULLAIN等人利用193nm抽运光抽运碘甲烷分子,再以CH3和I的共振波长的探测激光探测离子研究了碘甲烷分子的预解离动力学过程[18]。
-
飞秒时间分辨质谱实验中,利用采集程序记录不同离子的信号强度随延迟时间的依赖关系,可以获得不同抽运-探测时间延迟下的离子质谱,即不同离子的时间分辨质谱。用指数函数和仪器响应函数的卷积对时间分辨质谱进行拟合,能得到激发态和里德堡态的准确寿命。目前,利用飞秒时间分辨质谱对电子激发态寿命的测量已经由分子体系的研究向自由基体系拓展开来。
中国科学院ZHANG小组利用飞秒时间分辨质谱技术,测得了3-甲基吡啶分子里德堡态的准确寿命,用400nm两个光子吸收的方法布居了3-甲基吡啶的3s里德堡态。研究表明,3s里德堡态的寿命为62fs[19]。
飞秒时间分辨质谱技术特别是在测量寿命极短的分子或自由基的寿命中发挥非常重要的作用。例如,德国FISCHER课题组对活性很高、寿命极短的卡宾自由基激发态的寿命进行测量。在实验中,他们首先在实验室生成气相卡宾,然后利用飞秒时间分辨质谱技术[20]准确测得了寿命极短的卡宾自由基激发态寿命,如图 4所示。图 4中,m/z表示荷质比,m表示带电体的质量,z表示带电体的电荷量。

图 4 利用飞秒时间分辨质谱技术研究卡宾自由基激发态寿命
-
分子电子激发态超快异构化过程的研究中,飞秒时间分辨质谱技术也发挥着非常重要的作用,被证明为研究异构化过程的非常有效的手段。
飞秒时间分辨质谱技术在电子激发态超快异构化过程中应用的典型工作是FUSS等人对1, 3-环己二烯等分子体系的开环反应[21-22]进行了详细的研究。实验中他们用一束合适波长的飞秒抽运光把母体分子选择性的抽运到第一电子激发态上,分子激发到激发态后会通过不同衰减通道进行衰减,而1, 3-环己二烯分子被激发到第一激发态后发生超快异构化过程进行衰减,选择合适波长的另外一束飞秒探测激光在不同的抽运探测延迟时间跟1, 3-环己二烯分子发生作用,使被激发的母体分子电离,将这个异构化过程反映到实验上可观测态的离子态上。通过研究分析一系列实验中测得的离子信号随着抽运-探测时间演化的时间分辨质谱,能够得到1, 3-环己二烯分子被激发到第一电子激发态后势能发生的变化,并通过开环异构过程进行衰减,如图 5所示。图中,hνpu表示光子能量,h为普朗克常数,νpu为辐射电磁波的频率。
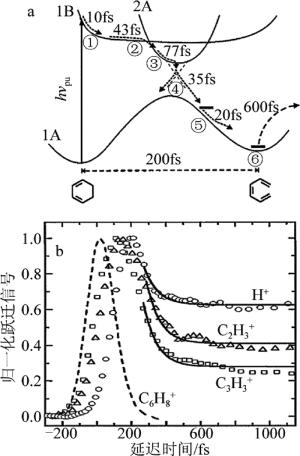
图 5 利用飞秒时间分辨质谱技术研究1,3-环己二烯的开环反应机理
-
利用飞秒时间分辨质谱技术可以测量激发态布居数随时间演化。根据分子电子激发态布居数随时间演化信息, 可以反推出分子激发态势能面之间的非绝热耦合信息,因此,飞秒时间分辨质谱技术也可以有效地探测超快内转换过程[23]。
西班牙LONGARTE小组利用283nm~243nm的可调谐激光将吲哚分子抽运到La带,被激发的吲哚分子在不同抽运探测时间延迟下被305nm的探测光电离,他们采集了不同抽运波长不同抽运探测时间延迟下的质谱。对不同抽运探测时间质谱信号拟合得到的数据进行分析可以推断吲哚分子La电子激发势能曲线的耦合信息[24]。吲哚分子的时间分辨质谱和势能曲线耦合信息,揭示了吲哚分子的内转换动力学过程。
中国科学院ZHANG研究员小组利用400nm抽运激光、800nm探测激光研究了C6H4(CH3)2[25]和BzCl[26]等分子体系激发态超快内转换动力学过程。
最近日本京都大学SUZUKI教授小组用飞秒时间分辨质谱技术和飞秒时间分辨光电子影像技术研究了吡嗪分子的激发态动力学[27],他们用抽运光激发吡嗪分子S2激发态,利用另外一束飞秒空紫外线(vacuum ultraviolet, VUV)光作为探测光观察到了S2/S1和S1/S0之间的内转换动力学过程,由于他们这次把飞秒VUV的光作为探测光,更清楚地观察到了把飞秒紫外光(ultraviolet,UV)作为探测光无法直接观察到的态。同时,他们提出将时间分辨质谱技术、光电子影像技术和液体束技术相结合的方法有着很大潜力和实用价值,势必会被广泛地应用到液相相关的物理、生物、化学等领域的科学研究。
-
在多重度不同的态之间的耦合过程(系间交叉过程)的研究中,时间分辨质谱技术发挥了非常重要的作用。
SUZUKI教授小组用飞秒时间分辨质谱技术研究吡嗪分子S1态的非绝热动力学过程[28],他们通过飞秒时间分辨质谱技术观察到了S1态衰减的同时三重态T1态的布居,由此证明吡嗪分子S1态的衰减主要通过T1态的系间交叉过程来实现。
中国科学院ZHANG小组利用400nm抽运激光、800nm探测激光研究了苯分子激发态超快动力学过程,观察到了时间尺度为1.06ps的S2态和T3态之间的系间交叉过程[29]。
-
飞秒时间分辨质谱技术和VUV的超短脉冲激光光源相结合可以非常有效地探测分子里德堡态超快动力学过程[30]。FARMANARA等人用VUV超短激光脉冲对CH3CO,C3H2Cl和丙酮分子里德堡态的动力学过程[31]进行研究。他们通过扫描不同抽运探测时间延迟下的母体离子和碎片离子信号,分析母体离子和碎片离子信号随时间变化,得到了这些分子里德堡态超快动力学信息。
亚利桑那大学的SANDHU[32]课题组开展了超激发态的氧分子里德堡态等力学过程的研究。他们把氧分子激发到超激发态,然后利用800nm的探测光探测超激发态的自电离和预解离过程。实验中发现,主量子数较低的里德堡态主要发生自电离过程。
日本京都大学SUZUKI教授小组利用脉宽小于20fm的深紫外光和VUV光作为抽运光和探测光,用飞秒时间分辨质谱技术和光电子影像技术研究了衰减寿命极短的里德堡态的动力学过程[33]。
-
飞秒时间分辨质谱技术原理简单,是飞秒抽运-探测技术与飞行时间质谱技术的结合,利用抽运光使分子被激发,制备随时间演化的量子波包;利用探测光在不同时间延迟下与分子发生作用,探测激发态量子波包的演化,记录不同抽运探测时间延迟的离子质谱。因此,飞秒时间分辨质谱技术已经在实时观察解离过程、测量分子电子激发态、里德堡态准确寿命、观察超快内转换、异构化等非绝热耦合过程等分子电子激发态超快无辐射动力学的研究中发挥着很重要的作用。由于飞秒时间分辨质谱技术可以实现对激发态寿命的直接测量、激发态动力学的实时探测、过渡态的实时跟踪,将会在构象动力学、匀共轭相互作用等新现象的研究当中发挥很重要的作用。
飞秒时间分辨质谱技术在超快动力学中的应用进展
Technology of femtosecond time resolution mass spectroscopy and its applications in ultrafast dynamics
-
摘要: 飞秒时间分辨质谱技术是飞秒抽运-探测技术与飞行时间质谱技术的结合。可以测得在不同抽运-探测时间延迟下,分子电子激发态电离或解离而来的离子质谱; 不同抽运-探测时间延迟下,质谱信号强弱的变化反映了激发态布居数的时态信息; 给出了分子激发态和里德堡态中准确的寿命信息、分子激发态势能面非绝热耦合信息以及分子过渡态信息。介绍了飞秒时间分辨质谱技术在分子激发态研究中的最新应用进展,以及在里德堡态解离、异构化、内转换、系间交叉等超快动力学过程研究中的最新进展。指出飞秒时间分辨质谱技术将在一些新现象的研究中发挥重要的作用。Abstract: Femtosecond time-resolved mass spectrometry is the combination of femtosecond pump-probe technique and flight time mass spectrometry. By the technology, ion mass spectra from ionization or dissociation of the molecules excited state can be measured under different pump-probe time delays. The temporal information of the population of the excited states varies with the change of the strength of mass spectra. Some information is given, such as:the accurate lifetime of the excited state and Rydberg state, coupling information of potential energy surface and non adiabatic of molecular excited state, and excessive state information. The application of femtosecond time-resolved mass spectrometry in ultrafast dynamics process is introduced, such as:recent advances of molecular excited states and the latest progress of Rydberg state for dissociation, isomerization, conversion, and system cross. And it is pointed out that femtosecond time-resolved mass spectrometry will play an important role in the study of some new phenomena.
-
Key words:
- laser technique /
- ultrafast dynamics /
- mass spectroscopy /
- femtosecond time resolution
-
[1] EYRING H. The activated complex in chemical reactions. Journal of Chemical Physics, 1934, 3(2):107-115. [2] ZEWAIL A H. Femtochemistry: Atomic-scale dynamics of the chemical bond. The Journal of Physical Chemistry, 2000, A104(24): 5660-5694. [3] KHUNDKAR L R, ZEWAIL A H. Picosecond mpi mass spectrometry of CH3I in the process of dissociation. Chemical Physics Letters, 1987, 142(6): 426-432. [4] CORRALES M E, LORIOT V, ZEWAIL A H. Structural dynamics effects on the ultrafast chemical bond cleavage of a photodissociation reaction. Physical Chemistry Chemical Physics, 2013, 16(19):8812-8818. [5] YU H. Study on measurement of femtosecond laser pulse width. Laser Technology, 2013, 37(5):679-681(in Chinese). [6] YANG M H, JIN Q, LIU J S, et al. CO molecular orientation controlled by combination of chirped THz pulse and femtosecond laser pulse. Laser Technology, 2015, 39(6):735-740(in Chinese). [7] VALDMANIS J A, FORK R L, GORDON J P.Generation of optical pulse as short as 27 femtoseconds directly from alaser balancing self -phase modulati on group -velocity dispersion saturable absorption, and saturablegain.Optics Letters, 1985, 10(3):131-133. [8] HE Y X, MU B L, LI J, et al.Relationship between Gaussian beam quality and wavefront aberration. Laser Technology, 2014, 38(6):747-752(in Chinese). [9] LUDOWISE P, BLACKWELL M, CHEN Y. Femtosecond time-resolved mass and photoelectron spectroscopic study of OC10 photodissociation. Coherent energy transfer in a stepwise reaction. Chemical Physics Letters, 1997, 273(3/4):211-218. [10] HO J W, CHEN W K, CHENG P Y. A direct observation of a concerted two-bond breaking reaction. Journal of the American Chemical Society, 2007, 129(25): 3748-3785. [11] HO J W, CHEN W K, CHENG P Y. Unraveling complex three-body photodissociation dynamics of dimethyl sulfoxide: a femtosecond time-resolved spectroscopic study. Journal of Physical Chemistry, 2008, A112(40): 10453-10468. [12] CHEN W K, HO J W, CHENG P Y. Ultrafast photodissociation dynamics of acetone at 195nm: Ⅱ initial state, intermediate, and product temporal evolutions by femtosecond mass-selected multiphoton ionization spectroscopy. Journal of Physical Chemistry, 2005, A109(34): 6805-6817. [13] CHEN W K, CHENG P Y. Ultrafast photodissociation dynamics of acetone at 195nm: Ⅱ unraveling complex three-body dissociation dynamics by femtosecond time-resolved photofragment translational spectroscopy. Journal of Physical Chemistry, 2005, A109(34): 6818-6829. [14] GITZINGER G, CORRALES M E, LORIOT V. A femtosecond velocity map imaging study on B-band predissociation in CH3I. Ⅰ. The band origin. The Journal of Chemical Physics, 2010, 132(23): 234313. doi: 10.1063/1.3455207 [15] ZHANG R R, SHEN H, QIN C C, et al. Ultrafast dissociation dynamics of acrylic acid studied with femtosecond pump-probe technique. Acta Physico-Chimica Sinica, 2012, 28(3):522-527. [16] XU Y Q, QIU X J, ABULIMITI B, et al. Energy transfer of ethyl iodine studied by time-resolved photoelectron imaging. Chemical Physics Letters, 2012, 554(12):53-56. [17] LIU Zh M, WANG Y M, ZHANG B, et al. Photodissociation dynamics of 2-iodotoluene investigated by femtosecond time-resolved mass spectrometry. Chinese Journal of Chemical Physics, 2016, 29(1):53-58. [18] POULLAIN S M, SAMARTZIS P C, KITSOPOULOS T N. New insights into the photodissociation of methyl iodide at 193nm: stereodynamics and productbranching ratios. Physical Chemistry Chemical Physics, 2015, 17(44): 29958-29968. doi: 10.1039/C5CP04850H [19] ABULIMITI B, QIU X J, DING Z H, et al. Studies on ultrafast dynamic of 3-picoline with femtosecond time-resolved photoelectron imaging. Acta Physico-Chimica Sinica, 2014, 30(1): 22-27. [20] NOLLER B, POISSON L, MAKSIMENKA R, et al. Femtosecond dynamics of isolated phenylcarbenes. Journal of the American Chemical Society, 2008, 130(45): 14908-14909. doi: 10.1021/ja804133c [21] FUSS W, SCHMID W E, TRUSHIN S A. Ultrafast dynamics of cyclohexene and cyclohexene-d10 excited at 200nm. Journal of the American Chemical Society, 2001, 123(33): 7101-7108. [22] FUSS W, SCHMID W E, TRUSHIN S A. Time-resolved dissociative intense-laser field ionization for probing dynamics: Femtosecond photochemical ring opening of 1, 3-cyclohexadiene. The Journal of Chemical Physics, 2000, 112(19): 8347-8362. doi: 10.1063/1.481478 [23] WU G R, NEVILLE S P, SCHALK O. Excited state non-adiabatic dynamics of pyrrole: A time-resolved photoelectron spectroscopy and quantum dynamics study. The Journal of Chemical Physics, 2015, 142(7): 074302. doi: 10.1063/1.4907529 [24] MONTERO R, CONDE A P, OVEJAS V, et al. Ultrafastphotophysics of the isolated indole molecule.Journal of Physical Chemistry, 2012, A116(11): 2698-2703. [25] LIU Y Z, TANG B F, SHEN H, et al. Probing ultrafast internal conversion of o-xylene via femtosecond time-resolved photoelectron imaging. Optics Express, 2010, 18(10): 5791-5801. [26] DING Z H, QIU X J, XU Y Q, et al.Ultrafast internal conversion dynamics of benzyl chloride by femtosecond time-resolved photoelectron imaging. Acta Physico-Chimica Sinica, 2012, 28(12): 2761-2766. [27] HORIO T, SPESYVTSEVJ R, SUZUKI T. Full observation of ultrafast cascaded radiationless transitions from S2 state of pyrazine using vacuum ultraviolet photoelectron imaging.The Journal of Chemical Physics, 2016, 145(4): 044306. [28] SUZUKI T, WANG L, KOHGUCHI H. Femtosecond time-resolved photoelectron imaging on ultrafast electronic dephasing in an isolated molecule. The Journal of Chemical Physics, 1999, 111(11): 4859-4861. doi: 10.1063/1.479822 [29] QIU X J, QIN C C, WANG J, et al.Direct imaging of the electronic dephasing in benzene: Experimental evidence for ultrafast intersystem crossing of T3←S2 states. Physical Review, 2012, A86(3): 032505. [30] LUCAS M, LIU Y, BRYANT R, et al. Vacuum ultraviolet photodissociation dynamics of methanol at 121.6. Chemical Physics Letters, 2015, 619(1):18-22. [31] FARMANARA P, STERT V, RADLOFF W. Ultrafast photodissociation dynamics of acetone excited by femtosecond 155nm laser pulses. Chemical Physics Letters, 2000, 320(6): 697-702. [32] TIMMERS H, SHIVARAM N, SANDHU A. Ultrafast dynamics of neutral superexcited oxygen: A direct measurement of the competition on between autoionization and predissociation. Physical Review Letters, 2012, 109(17): 173001. doi: 10.1103/PhysRevLett.109.173001 [33] SPESYVTSEVJ R, HORIO T, SUZUKI T. Excited-state dynamics of furan studied by sub-20fs time-resolved photoelectron imaging using 159nm pulses. The Journal of Chemical Physics, 2015, 143(1): 014302. -
 点击查看大图
点击查看大图

图(5)
计量
- 文章访问数: 3307
- HTML全文浏览量: 1959
- PDF下载量: 17
- 被引次数: 0
